Analyze neutrophil counts of NARES data
Sehyun Oh
March 24, 2022
Source:vignettes/NARES/neutrophil_counts_NARES.Rmd
neutrophil_counts_NARES.RmdAbstract
Source CodeSetup
In this vignette, we benchmarked Figure 3D of the MultiPLIER paper: Neutrophil count of NARES samples were estimated by MCPcounter and plotted against RAV1551-assigned sample scores.
Load packages
if (!"GenomicSuperSignaturePaper" %in% installed.packages())
devtools::install_github("shbrief/GenomicSuperSignaturePaper")
suppressPackageStartupMessages({
library(GenomicSuperSignature)
library(GenomicSuperSignaturePaper)
library(dplyr)
library(RColorBrewer)
library(ComplexHeatmap)
})RAVmodel
To directly compare our new analysis with the results from MultiPLIER paper, we used the RAVmodel annotated with the same priors as MultiPLER: bloodCellMarkersIRISDMAP, svmMarkers, and canonicalPathways.
RAVmodel <- getModel("PLIERpriors", load=TRUE)
RAVmodel
#> class: PCAGenomicSignatures
#> dim: 13934 4764
#> metadata(8): cluster size ... version geneSets
#> assays(1): RAVindex
#> rownames(13934): CASKIN1 DDX3Y ... CTC-457E21.9 AC007966.1
#> rowData names(0):
#> colnames(4764): RAV1 RAV2 ... RAV4763 RAV4764
#> colData names(4): RAV studies silhouetteWidth gsea
#> trainingData(2): PCAsummary MeSH
#> trainingData names(536): DRP000987 SRP059172 ... SRP164913 SRP188526
version(RAVmodel)
#> [1] "1.1.1"NARES Nasal Brushings
Expression data
NARES (Grayson et al., 2015) is a nasal brushing microarray dataset that includes patients with ANCA-associated vasculitis, patients with sarcoidosis and healthy controls among other groups. In the MultiPLIER paper, NARES data was projected into the MultiPLIER latent space.
exprs <- readr::read_tsv("data/NARES_SCANfast_ComBat_with_GeneSymbol.pcl") %>%
as.data.frame
rownames(exprs) <- exprs$GeneSymbol
dataset <- as.matrix(exprs[,3:ncol(exprs)])
dataset[1:2, 1:4]
#> N1004 N1007 N1017 N1025
#> A1BG 0.05641348 0.06333364 0.001833597 -0.05897817
#> NAT2 -0.14980276 -0.09186388 -0.093684629 -0.06080664MCPcounter
NARES data doesn’t have any information on the cell type composition of the samples. So in the MultiPLIER paper, the authors applied MCPcounter) (Becht et al., Genome Biology, 2016) , a method for estimating cell type abundance in solid tissues. As MultiPLIER paper, we tested whether the neutrophil-associated RAV is well-correlated with the neutrophil estimates of the NARES dataset.
* Install MCPcounter
devtools::install_github("ebecht/MCPcounter",
ref = "a79614eee002c88c64725d69140c7653e7c379b4",
subdir = "Source",
dependencies = TRUE)- Run MCPcounter
mcp.results <- MCPcounter::MCPcounter.estimate(exprs.mat,
featuresType = "HUGO_symbols")
mcp.melt <- reshape2::melt(mcp.results,
varnames = c("Cell_type", "Sample"),
value.name = "MCP_estimate")
readr::write_tsv(mcp.melt, "data/NARES_ComBat_MCPCounter_results_tidy.tsv")Transfer Learning
Validation
RAV1551, a neutrophil-associated RAV based on SLE-WB dataset, is not found in the top 5 validated RAVs of NARES dataset, implying that neutrophil count is not the major feature of NARES dataset.
val_all <- validate(dataset, RAVmodel)
validated_ind <- validatedSignatures(val_all, RAVmodel,
num.out = 5, swCutoff = 0,
indexOnly = TRUE)
heatmapTable(val_all, RAVmodel, num.out = 5, swCutoff = 0)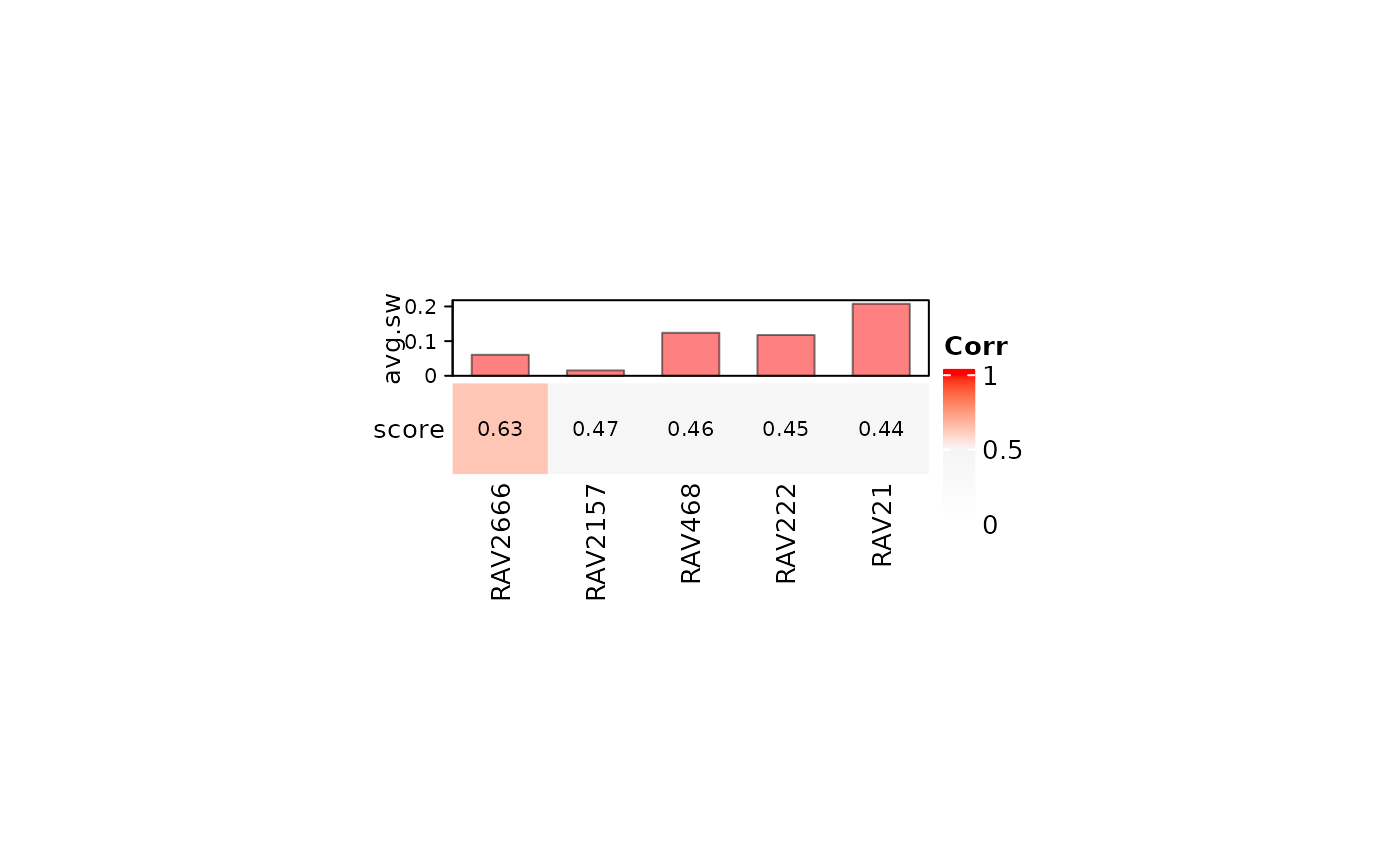
RAV1551 is actually ranked as the top 14th RAV based on its validation score.
r-squared value
sampleScore <- calculateScore(dataset, RAVmodel)
## Remove Sample category "gene"
gene_ind <- which(mcp.melt$Sample == "Gene")
mcp.melt <- mcp.melt[-gene_ind,]
## Subset sampleScore to join with MCPcounter
sampleScore_sub <- sampleScore[, 1551] %>% as.data.frame
sampleScore_sub <- tibble::rownames_to_column(sampleScore_sub)
colnames(sampleScore_sub) <- c("Sample", "RAV1551")
## Join with MCPcounter neutrophil estimates
dat <- dplyr::filter(mcp.melt, Cell_type == "Neutrophils") %>%
dplyr::inner_join(y = sampleScore_sub, by = "Sample")
head(dat, 3)
#> Cell_type Sample MCP_estimate RAV1551
#> 1 Neutrophils N1004 0.6064648 -0.09409653
#> 2 Neutrophils N1007 1.8775036 1.91008170
#> 3 Neutrophils N1017 0.6006657 -0.92446139We plot the neutrophil estimates against RAV1551-assigned sample scores.
plot <- LVScatter(dat, "RAV1551",
y.var = "MCP_estimate",
ylab = "MCPcounter Neutrophil Estimate",
title = "RAVmodel",
subtitle = "NARES Nasal Brushings")
plot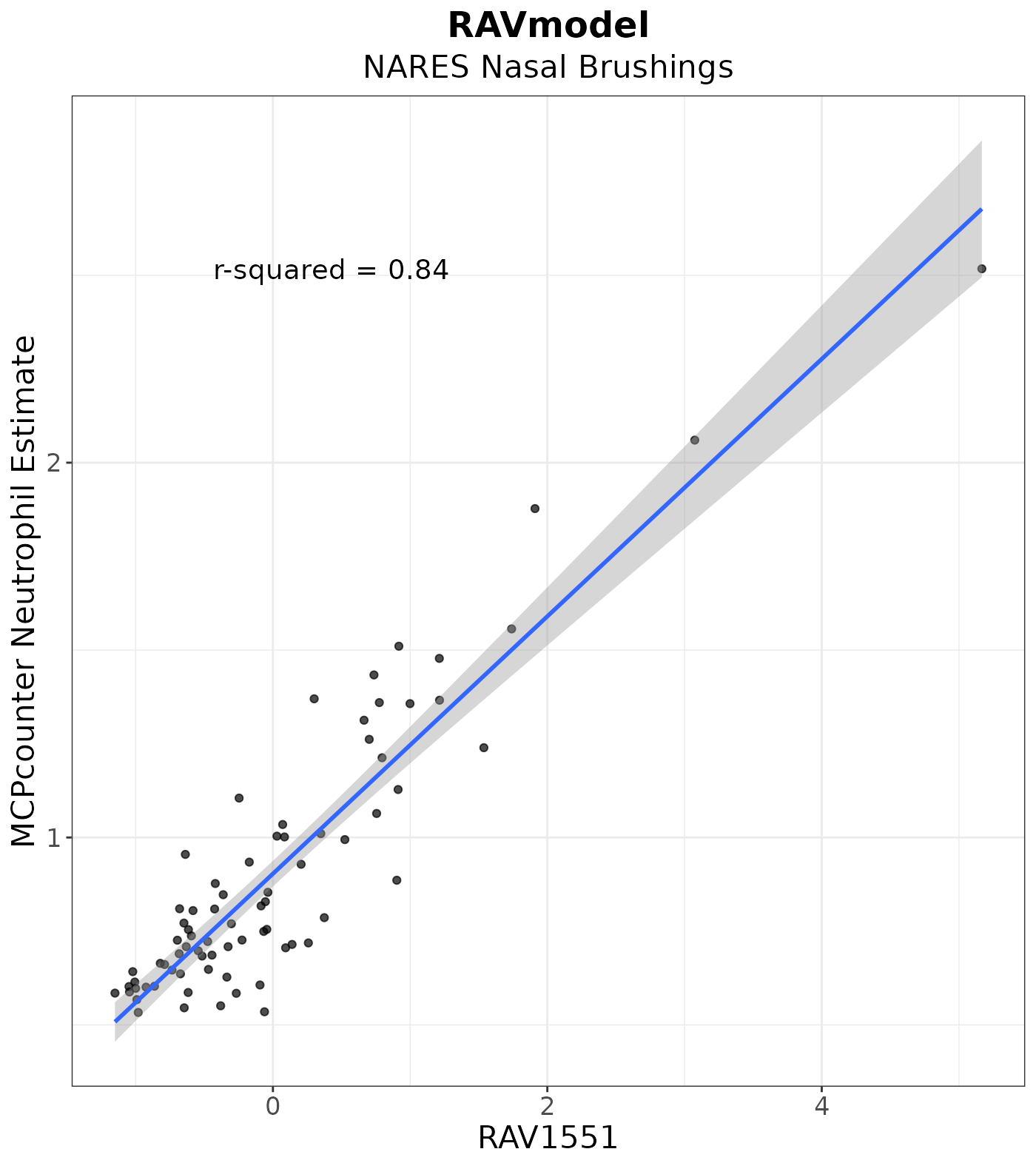
Session Info
r sessionInfo() #> R version 4.1.2 (2021-11-01) #> Platform: x86_64-pc-linux-gnu (64-bit) #> Running under: Ubuntu 20.04.3 LTS #> #> Matrix products: default #> BLAS/LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.8.so #> #> locale: #> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C #> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8 #> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8 #> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C #> [9] LC_ADDRESS=C LC_TELEPHONE=C #> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C #> #> attached base packages: #> [1] grid stats4 stats graphics grDevices utils datasets #> [8] methods base #> #> other attached packages: #> [1] ComplexHeatmap_2.10.0 RColorBrewer_1.1-2 #> [3] dplyr_1.0.8 GenomicSuperSignaturePaper_1.1.1 #> [5] GenomicSuperSignature_1.3.6 SummarizedExperiment_1.24.0 #> [7] Biobase_2.54.0 GenomicRanges_1.46.1 #> [9] GenomeInfoDb_1.30.1 IRanges_2.28.0 #> [11] S4Vectors_0.32.3 BiocGenerics_0.40.0 #> [13] MatrixGenerics_1.6.0 matrixStats_0.61.0 #> [15] BiocStyle_2.22.0 #> #> loaded via a namespace (and not attached): #> [1] colorspace_2.0-3 ggsignif_0.6.3 rjson_0.2.21 #> [4] ellipsis_0.3.2 rprojroot_2.0.2 circlize_0.4.14 #> [7] XVector_0.34.0 GlobalOptions_0.1.2 fs_1.5.2 #> [10] clue_0.3-60 farver_2.1.0 ggpubr_0.4.0 #> [13] bit64_4.0.5 fansi_1.0.2 splines_4.1.2 #> [16] codetools_0.2-18 doParallel_1.0.17 cachem_1.0.6 #> [19] knitr_1.37 jsonlite_1.8.0 broom_0.7.12 #> [22] cluster_2.1.2 dbplyr_2.1.1 png_0.1-7 #> [25] BiocManager_1.30.16 readr_2.1.2 compiler_4.1.2 #> [28] httr_1.4.2 backports_1.4.1 assertthat_0.2.1 #> [31] Matrix_1.4-0 fastmap_1.1.0 cli_3.2.0 #> [34] htmltools_0.5.2 tools_4.1.2 gtable_0.3.0 #> [37] glue_1.6.2 GenomeInfoDbData_1.2.7 rappdirs_0.3.3 #> [40] Rcpp_1.0.8.3 carData_3.0-5 jquerylib_0.1.4 #> [43] pkgdown_2.0.2 vctrs_0.3.8 nlme_3.1-155 #> [46] iterators_1.0.14 xfun_0.30 stringr_1.4.0 #> [49] lifecycle_1.0.1 rstatix_0.7.0 zlibbioc_1.40.0 #> [52] scales_1.1.1 vroom_1.5.7 ragg_1.2.2 #> [55] hms_1.1.1 parallel_4.1.2 yaml_2.3.5 #> [58] curl_4.3.2 memoise_2.0.1 ggplot2_3.3.5 #> [61] sass_0.4.0 stringi_1.7.6 RSQLite_2.2.10 #> [64] highr_0.9 desc_1.4.1 foreach_1.5.2 #> [67] filelock_1.0.2 shape_1.4.6 rlang_1.0.2 #> [70] pkgconfig_2.0.3 systemfonts_1.0.4 bitops_1.0-7 #> [73] evaluate_0.15 lattice_0.20-45 purrr_0.3.4 #> [76] labeling_0.4.2 bit_4.0.4 tidyselect_1.1.2 #> [79] magrittr_2.0.2 bookdown_0.25 R6_2.5.1 #> [82] magick_2.7.3 generics_0.1.2 DelayedArray_0.20.0 #> [85] DBI_1.1.2 pillar_1.7.0 mgcv_1.8-38 #> [88] abind_1.4-5 RCurl_1.98-1.6 tibble_3.1.6 #> [91] crayon_1.5.0 car_3.0-12 utf8_1.2.2 #> [94] BiocFileCache_2.2.1 tzdb_0.2.0 rmarkdown_2.13 #> [97] GetoptLong_1.0.5 blob_1.2.2 digest_0.6.29 #> [100] tidyr_1.2.0 textshaping_0.3.6 munsell_0.5.0 #> [103] bslib_0.3.1