Research
Oh Lab’s Focus
Database Search and Transfer Learning

We developed a method for interpreting new transcriptomic datasets through instant comparison to public datasets without high-performance computing requirements. This method consists of a pre-computed model and an out-of-the-box Bioconductor software package, GenomicSuperSignature, which applies the model to new data. Using human datasets, we demonstrated our method’s efficient and coherent database search capabilities, robustness to batch effects and heterogeneous training data, and transfer learning capacity. We are currently expanding this method to different species (e.g., mouse, yeast) and different omics data types (e.g., scRNAseq, metagenomics). Additionally, leveraging the transfer learning capacity of this method, we are developing a systematic framework for inferring various phenotypes from transcriptomic data.
Cross-study Metadata Standardization

To enhance the FAIRness of public omics data, we aim to systematically harmonize and standardize metadata across major public biomedical data repositories. We are performing large-scale manual metadata curation for publicly available omics data while building an automated metadata harmonization pipeline that leverages various Natural Language Processing (NLP) techniques, including Large Language Models (LLMs), using manually harmonized metadata as a gold standard. The standardization process integrates ontologies, a data reconciliation system, and scientific provenance tracking to ensure the interpretability, accuracy, and reliability of the harmonized data. Currently, we are targeting several major public omics data repositories for standardization. The outcome of this project will transform how researchers discover and integrate public data into new research topics, ultimately accelerating biological discovery.
Collaborations
Histopathology Image Data Analysis
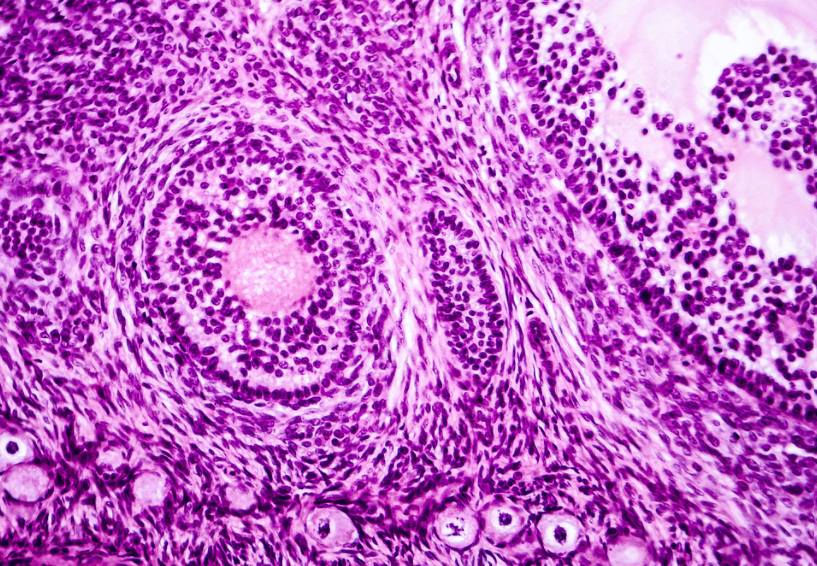
Histopathological images provide unparalleled insights into tissue architecture, cellular morphology, and tumor spatial organization. However, analyzing these images often requires non-R software, unlike other high-throughput omics data and downstream statistical analyses that are frequently performed in the R/Bioconductor environment. A seamless connection between these platforms is critical for integrative multi-modal analyses that combine both omics and image data. To address this gap, we are creating standardized workflows that process raw image files and extract image features accessible in R, while also developing a comprehensive repository of image features extracted from The Cancer Genome Atlas (TCGA) images.
curatedMetagenomicData v4
We are leading the metadata curation effort for curatedMetagenomicData (cMD) version 4, a major update to the widely used Bioconductor resource of uniformly processed human microbiome data. cMD v4 transitions the sample-level metadata to a new FAIR-compliant schema that incorporates standardized ontology terms, a data dictionary, and automated validation to ensure interpretability and reproducibility across studies. We orchestrate the end-to-end curation pipeline — including initiation, validation, and deployment — and provide tooling (e.g., makeCombinedMetadata, validateStudy) that enables community contributors to add new studies. This work substantially expands the harmonized sample collection available to the research community and lays the foundation for downstream meta-analyses of the human microbiome across disease, treatment, and population contexts.